Main
Alcohol use disorder (AUD) is characterized by unhealthy alcohol use and includes multiple clinical symptoms, such as the loss of control over alcohol intake, the development of tolerance, and withdrawal symptoms when alcohol is not consumed1. AUD is a heterogeneous phenotype, with affected individuals exhibiting a wide variety of alcohol use behaviors that are associated with different clinical, social and economic correlates2,3. At the same time, AUD is a common disorder that affects about 10% of the population in a given year and is associated with substantial morbidity and mortality4. Despite the negative health effects, treatment options for AUD are limited, and there is no clear understanding of the underlying neurobiology5.
Recently, there has been an increasing interest in how epigenetic factors contribute to the risk and pathology of AUD6,7. Epigenetic modifications are reversible changes that can influence gene function without altering the DNA sequence itself, thus serving as vital and potentially mechanistically informative genetic and environmental indicators of gene–environment interaction8. This is particularly relevant for AUD, for which the heritability is estimated to be about 50% on the basis of twin studies9. While genome-wide association studies (GWASs) can reflect the risk of developing an AUD, epigenome-wide association studies (EWASs) additionally capture the effects of alcohol exposure, next to other environmental and lifestyle factors, and could contribute to disentangling the heterogeneity in AUD. Epigenetic modifications involve various mechanisms, with DNA methylation being the most studied in humans. DNA methylation primarily involves the addition of a methyl group to the 5′ position of cytosines and is typically observed in cytosine–guanine dinucleotides (CpG sites)8. DNA methylation is highly tissue- and cell type-specific. While it is metabolized primarily in the liver, alcohol has systemic effects on the entire organism10. Because of the tissue specificity of DNA methylation in combination with the widespread effects of alcohol, investigating DNA methylation in peripheral blood offers the opportunity to capture these systemic effects. At the same time, there is high translational value in investigating peripheral DNA methylation, which allows for the construction of methylation scores that have potential as biomarkers, as demonstrated for aging11, alcohol consumption12 and smoking13. A similar marker, based on robust DNA methylation associations, could plausibly be constructed for AUD14.
Several EWASs have investigated the association between DNA methylation and AUD phenotypes in peripheral blood samples15,16,17. However, small sample sizes ranging between 198 and 1,132, and the resulting limited statistical power, have produced inconsistent results—but also provided the basis for a larger investigation across cohorts. So far, replicated CpG sites have been observed in GAS5, which was identified as a target for epigenetic modification in an EWAS comparing 379 individuals with and 246 without AUD in the National Institute on Alcohol Abuse and Alcoholism (NIAAA) cohort15. This site was also replicated in an independent sample, the Grady Trauma Project (GTP). The largest EWAS to date included 323 individuals with AUD and 809 unaffected controls in the Netherlands Study of Depression and Anxiety (NESDA)16. Here DLGAP1 was identified and confirmed in 412 participants of the Great Smoky Mountains Study (GSMS)16. In addition to the need to increase sample sizes, it is important to investigate DNA methylation associated with AUD across heterogeneous cohorts to identify robust signals representative of the heterogeneity of the phenotype regarding severity, comorbidities and somatic factors, among others. The currently available studies range from cohorts ascertained for AUD to population-based approaches and cohorts originally ascertained for comorbid disorders, such as major depressive disorder (MDD) and post-traumatic stress disorder (PTSD). Large-scale meta-analyses or large samples are necessary to facilitate valid and replicable results that are independent of specific cohort profiles. The advantages of meta-analysis are increasing the sample size, enhancing statistical power and including cohorts with different characteristics; a disadvantage is heterogeneity that may be hard to identify or account for statistically.
This study aimed to identify DNA methylation signals robustly associated with AUD, construct a methylation risk score (MRS) and perform out-of-sample predictions. To accomplish this, we first conducted a large meta-analysis of EWASs on AUD, investigating 7 cohorts with a total sample size of 3,775 individuals (AUD = 1,325, controls = 2,450). Next we carried out extensive downstream analyses, including overrepresentation analysis of gene ontology terms and genes identified in GWASs to explore potential functional mechanisms behind the findings. We also tested the overlap with alcohol consumption and the blood–brain concordance and performed sensitivity analyses to control for smoking effects. Finally, we constructed an MRS and validated it in an independent cohort (N = 2,534).
Results
Meta-analysis of AUD
To identify robust associations of DNA methylation and AUD, a meta-analysis of standardized, cohort-level EWASs was performed, resulting in a sample size of 3,775 individuals of primarily European Ancestry (Table 1 and Supplementary Table 1). Participating cohorts included 194 male patients with AUD, recruited at the Central Institute of Mental Health (CIMH) in Mannheim17, 231 participants from the Yale–Penn cohort18,19, 442 participants from the University of California at San Francisco (UCSF) Family Alcoholism Study20, 677 individuals who participated in the GTP21, 615 patients who were treated at the NIAAA15, 484 participants of the GSMS22,23 and 1,132 participants of the NESDA24,25. Detailed information on the cohorts can be found in Supplementary Text 1. The main model tested the association of DNA methylation with AUD, including covariates for sex, age, self-reported smoking, cell type composition and three genotype principal components to control for population stratification. All array-based EWASs also included four principal components of the internal control probes to control for batch effects. The quantile–quantile plot and lambda value (λ = 1.28) indicated a modest degree of inflation (Extended Data Fig. 1). Bacon correction was applied to control inflation, resulting in a final lambda of 1.06.
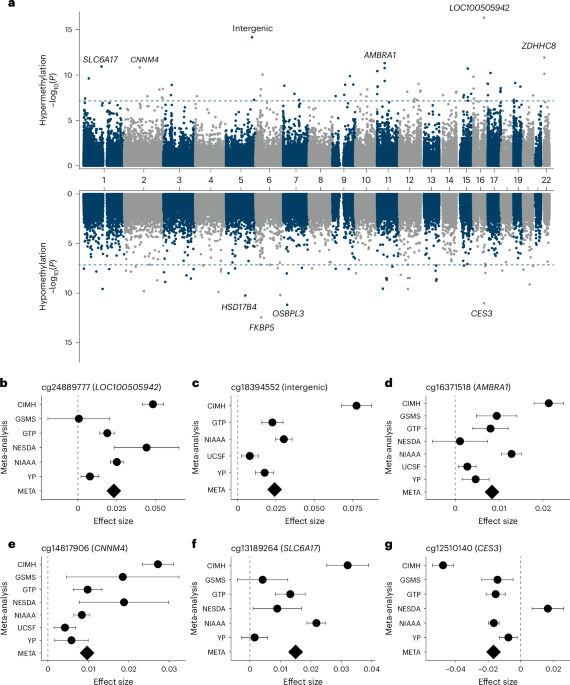
In the fixed-effects meta-analysis, 118 CpG sites were significantly associated with AUD after Bonferroni correction (Fig. 1 and Supplementary Table 2). Of the 118 CpG sites, 106 were also significant in the random-effects meta-analysis, supporting the robustness of the associations. Genome-wide CpG methylations are non-independent. However, overall patterns of covariance are not well understood and are likely to be affected by tissue, developmental timing and exposure. Thus, for the purposes of this analysis, a conservative threshold for significance, assuming independence of the CpGs assayed, was used. The strongest association was observed for cg24889777 (Bbacon = 0.021, where B denotes the effect and 0.021 represents a 2.1% methylation difference; Pbacon = 5.12 × 10−17) in the long non-coding RNA LOC100505942. The top CpG sites showed consistent effects across cohorts, which are depicted in Fig. 1b–g for the six most significant CpG sites exhibiting converging effects in at least five of the seven cohorts. Each of these was also directionally consistent.
a, DMPs split into hypermethylated (upper panel) and hypomethylated (lower panel) CpG sites; genes of the ten most significant DMPs are annotated; blue dashed line indicates epigenome-wide significance after Bonferroni correction (two-sided test). x axis depicts the location on the chromosome. b–g, Regression coefficients for AUD for the six most significant CpG sites (cg24889777 (b), cg18394552 (c), cg16371518 (d), cg14817906 (e), cg13189264 (f) and cg12510140 (g)) in the meta-analysis with converging effect estimates in at least five cohorts; dots represent unstandardized coefficients from the meta-analysis, error bars represent standard errors of estimate and diamond shape represents effect in meta-analysis. Blue dashed line indicates effect size in the meta-analysis. All tests have been performed two-sided. META, primary meta-analysis (N = 3,775); YP, Yale–Penn (N = 231); UCSF (N = 442); NIAAA (N = 615); NESDA (N = 1,132); GTP (N = 677); GSMS (N = 484); CIMH (N = 194).
To evaluate technical effects due to DNA methylation assessment, we conducted an additional analysis in which cohorts evaluating DNA methylation via microarray or methyl-binding-domain sequencing (MBD-seq) were meta-analyzed separately. This was followed by a second meta-analysis. The correlation between the effect sizes of the microarray analysis and the primary analysis was r = 0.99 (P < 2.2 × 10−16), while the correlation between the MBD-seq results and the primary analysis was r = 1 (P < 2.2 × 10−16; Extended Data Fig. 2a,b).
In addition, 57 significantly differentially methylated regions (DMRs) were identified after false discovery rate (FDR) correction (Supplementary Table 3 and Extended Data Fig. 2c). The top significant DMR was in HNRNPA1 (PFDR = 1.62 × 10−17), while the DMR containing the most significant CpG site was annotated in the AMBRA1 gene (PFDR = 1.43 × 10−15).
Gene ontology overrepresentation analysis
The 118 epigenome-wide significant CpG sites were enriched for biological processes related to amino acid transport, such as ‘L-leucine transport’ (GO:0015820; z = 14.26, PFDR = 0.016) and ‘branched-chain amino acid transport’ (GO:0015803; z = 11.98, PFDR = 0.023), molecular functions related to GTPase binding and transmembrane transporter activity, cellular components related to vesicle transport, such as ‘transport vesicle membrane’ (GO:0030658; z = 5.79, PFDR = 0.0063) and ‘phagocytic vesicle’ (GO:0045335; z = 6.94, PFDR = 0.0063), as well as membrane-related gene ontology (GO) terms. As with CpG sites, DMRs were enriched for membrane-related cellular components and molecular functions related to GTPase binding and transmembrane transporter activity. Enrichment was also observed for biological processes related to response to nutrient levels, such as ‘cellular response to starvation’ (z = 8.43, PFDR = 0.005). Results are visualized in Extended Data Fig. 3, and all nominally significant GO terms for CpG sites are summarized in Supplementary Tables 4 and 5 for DMRs.
GWAS enrichment analysis
To explore whether genetic variance contributed to the findings, we formed three gene sets consisting of CpG sites identified in the primary meta-analysis, CpG sites identified in the primary meta-analysis with known methylation quantitative trait loci26, and DMRs. While none of these gene sets was significantly enriched for GWAS signals of problematic alcohol use27, alcohol dependence28, drinks per week29, cigarettes per day30, MDD31 or PTSD32 (all P > 0.056; Supplementary Table 6), CpG sites in DMRs showed the strongest associations with GWAS signals for alcohol dependence28 (P = 0.056) and drinks per week29 (P = 0.073).
Overlap with alcohol consumption EWAS
Because alcohol consumption is a necessary prerequisite to AUD, we investigated whether the DNA methylation signatures of AUD were distinct or shared with those of alcohol consumption traits. Therefore, we performed an overlap analysis with the results of a recent EWAS of alcohol consumption in the Generation Scotland cohort33. Of the 118 epigenome-wide significant CpG sites identified in the primary analysis, 65 (55.1%) were also significantly associated with alcohol consumption, indicating a substantial overlap between DNA methylation signatures of alcohol consumption and AUD (P < 2.2 × 10−16; Fig. 2a). The direction of effect was the same for all overlapping CpG sites (Fig. 2b). Two of the top ten CpG sites were exclusively associated with AUD, namely, the top hit, cg24889777, and cg03546163 in FKBP5.
Blood–brain concordance
Due to the tissue specificity of DNA methylation, we investigated the correlation between the DNA methylation levels of the identified CpG sites in blood and brain tissue. First, we investigated whether a correlation between blood and brain DNA methylation levels of the 118 epigenome-wide significant CpG sites associated with AUD in the primary meta-analysis could be observed using BECon34. This analysis revealed information on blood–brain concordance for 38 CpG sites. The full results are provided in Extended Data Fig. 4. Six CpG sites exhibited strong correlations (0.49 ≤ r ≤ 0.60) in Brodmann Area 10, that is, cg06846976 in TSC2 and cg04406114 in POU2F2, in Brodmann Area 20, that is, cg14380013 in UNC119B and cg03546163 in FKBP5, or in Brodmann Area 7, that is, cg18059012 in PATL2 and cg00422488 in ANP32B.
In addition, we investigated whether the 118 CpG sites identified in the primary meta-analysis overlapped with results from two recent EWASs of AUD in postmortem human brain tissue from Zillich et al.35 and White et al.36. In the two studies, five brain regions that play a central role in the neurocircuitry of addiction were investigated: dorsolateral prefrontal cortex/Brodmann Area 935,36, ventral striatum/nucleus accumbens35,36, caudate nucleus35, putamen35 and anterior cingulate cortex35. There was no overlap between the CpG sites identified in the primary meta-analysis and the postmortem brain EWASs.
Smoking sensitivity analysis
Across cohorts, individuals with AUD exhibited higher smoking rates than controls (Supplementary Table 1). Thus, we performed a sensitivity analysis in non-smoking individuals only as an approximate method to disentangle the influence of this potential confounder. Results from the sensitivity analysis in nonsmokers largely overlapped with those of the primary analysis (Extended Data Fig. 5).
Leave-one-out analysis
To systematically investigate the heterogeneity among the studies, we conducted leave-one-out analyses. All tested CpG sites remained significant in the leave-one-out analyses (all P < 4.86 × 10−4), except one when the NIAAA cohort was excluded: cg11376147 (P = 0.11). Forest plots of the leave-one-out analysis for the top CpG sites showing consistent effects across cohorts are depicted in Extended Data Fig. 6, and results are listed in Supplementary Table 7.
MRS
For a potential clinical translation, we constructed an MRS based on the summary statistics of the primary meta-analysis and validated the score in the independent Ludwigshafen Risk and Cardiovascular Health Study (LURIC) cohort. The LURIC cohort was recruited between 1997 and 2000 and included 3,316 patients who were hospitalized for a coronary angiography37. For 2,534 of these patients, DNA methylation data and detailed information on alcohol consumption were available. Patients reported their mean daily intake of alcoholic beverages, which was then converted to grams of alcohol consumed per day as described in ref. 38. While AUD diagnosis was not assessed in LURIC, the cohort includes a large number of participants (37.05%) exhibiting heavy drinking according to NIAAA criteria (Fig. 3a)39.
a, Alcohol consumption in LURIC by heavy-drinking status: red, heavy drinking; blue, no heavy drinking. b, Prediction accuracy (R2) of the MRS of AUD for P-value thresholds. c, Alcohol consumption per day in grams, predicted by the MRS of AUD; blue represents the regression line, gray area indicates the 95% confidence band of the regression line. d, ROC curve for heavy drinking.
MRSs were constructed using the pruning and thresholding method proposed by Chen et al.40. Here methylation values were first combined into co-methylated regions and, on the basis of different P-value thresholds, lead CpG sites per region were identified. The AUD MRS was then constructed on the basis of the weights of the lead CpG sites. The P-value thresholding resulted in a P-value threshold of 5 × 10−7, as presented in Fig. 3b. The resulting AUD MRS comprised 204 CpG sites after pruning and explained 2.06% of the variance in alcohol consumption (g d−1) in the LURIC cohort. When investigating the MRS in linear regression models, controlling for sex, age and cell type composition, the AUD MRS remained predictive of alcohol consumption (B = 5.24, s.e. = 0.59, P < 2 × 10−16). In addition to the AUD MRS, sex, age, and CD8+ and CD4+ T-cells predicted daily alcohol consumption (Supplementary Table 8), with the overall model explaining 10.44% of the variance.
Using logistic regression, we found that the AUD MRS predicted heavy drinking (B = 6.77, s.e. = 1.33, P < 2 × 10−16), serving as a potential proxy for AUD. Sex and the CD8+ T-cell proportion were also significant predictors in the model, although with smaller effects (Supplementary Table 9 and Fig. 3c). The logistic regression model fit was compared with a model not including the AUD MRS using a likelihood ratio test, which favored the model including the AUD MRS (P = 2.86 × 10−7). Nagelkerke’s R2 showed that the model accounted for 10.48% of the variance in heavy drinking, similar to the variance accounted for in daily alcohol consumption, with an area under the receiver operating characteristic curve (AUC-ROC) of 0.657 (Fig. 3d).
Discussion
The present meta-analysis identified several differentially methylated CpG sites and regions robustly associated with AUD. Our results highlight CpG sites associated with AUD, as well as genes previously associated with alcohol consumption and related phenotypes, and reproduced findings from previous studies15,16,17,33. We also constructed an MRS of AUD that predicted alcohol consumption and heavy drinking in an independent cohort.
The most significant differentially methylated position (DMP) was cg24889777 in the locus LOC100505942, which transcribes an uncharacterized non-coding RNA. This CpG site was also associated in previous EWASs of smoking and chronic obstructive pulmonary disease, incident liver cirrhosis, chronic pain, prevalent ischemic heart disease, and incident type 2 diabetes41,42; phenotypes that show a strong genetic and phenotypic association and clinical overlap with AUD43,44,45,46. Differential methylation in cg24889777 has not previously been associated with AUD or alcohol consumption. The association with traits and disorders comorbid with AUD suggests that methylation differences in cg24889777 could reflect shared underlying mechanisms between these traits that could affect non-coding RNA regulation.
Several CpG sites previously linked to AUD in individual studies were also identified in the primary meta-analysis, underlining the robustness of associations. For example, SLC1A2, which encodes the glutamate transporter 1, was robustly associated in the meta-analysis and in previous analyses15,17. Other genes include HNRNPA1 and AMBRA1, both of which emerged in the positional and regional analyses. AMBRA1 encodes the autophagy and beclin 1 regulator 1, responsible for GTPase binding and ubiquitin–protein ligase binding activity. It is expressed in various tissues, including the brain. As with the top DMP, the CpG site cg14222701, located in the AMBRA1 gene, was differentially methylated in previous EWASs of type 2 diabetes, chronic pain, smoking, chronic obstructive pulmonary disease, lung cancer and ischemic heart disease41,42. In two studies, differential methylation in AMBRA1 was associated with AUD17 or alcohol consumption12. Together with the DCX (DDB1-CUL4-X-box) E3 ubiquitin–protein ligase, AMBRA1 assembles into a complex that plays a role in the cell cycle and autophagy47. The substrates targeted by the DCX(AMBRA1) complex were previously associated with alcohol consumption. One example is the CND gene coding for Cyclin D, a polyubiquitination target47. In one study, different CCNDs were differentially expressed in the hippocampus of people with AUD, revealing pathways responsible for cell cycle regulation or cancer signaling to be associated with AUD48. The autophagosomal marker beclin 1, another target of the DCX(AMBRA1) complex, was increased in association with alcohol exposure and is thought to play a role in ethanol-induced autophagy49.
The gene ontology overrepresentation analysis indicated that differentially methylated CpG sites were functionally related to amino acid and vesicle transport, among other biological processes and cellular components. Among the genes driving the association with amino acid transport is SLC7A5, which is involved in blood–brain barrier transport50 and could contribute to blood–brain barrier disruption in AUD51. DMRs were related to autophagy, including cellular starvation and mTOR signaling, which is associated with ethanol-related neurotoxicity52.
We observed a substantial overlap between DNA methylation signatures of AUD and those of alcohol consumption33. While alcohol consumption is a prerequisite of AUD, the considerable overlap could indicate that some of the observed DNA methylation signatures reflect the prolonged heavy alcohol consumption associated with AUD. In line with this, alcohol consumption and AUD also show both shared and distinct genetic architectures, although GWAS signals are thought to primarily reflect vulnerability, while EWASs also capture the consequences of exposure. At the same time, our results did not indicate a strong enrichment of the results of the primary meta-analysis for GWAS signals of problematic alcohol use, alcohol dependence or drinks per week, although trends were observed for DMR enrichment for the latter two traits. To disentangle the genetic and epigenetic signatures of AUD, both polygenic and methylation risk scores can prove useful. In the present study, the MRS derived from the primary analysis predicted heavy drinking in an independent cohort. The performance of the MRS was similar to or slightly higher than those recently reported for other psychiatric phenotypes, such as MDD (AUC-ROC = 0.53)53 or for a childhood trauma MRS predicting psychopathology 17 years later in early adulthood (0.54 ≤ AUC-ROC ≤ 0.63)23. Still, the predictive performance of the MRS is insufficient for clinical application. Previous studies that developed MRSs of AUD did not test them in independent samples, or the datasets used to construct the MRS were from small samples14. Here we construct an MRS predictive of alcohol consumption and heavy drinking as a binary phenotype. Due to the lack of available data, the performance of the MRS was not tested in an independent sample of individuals with and without an AUD diagnosis. In addition, the characteristics of the validation sample—older patients hospitalized for a coronary angiography—could affect the performance of the MRS, underlining the need for future work to test the MRS in cohorts ascertained for AUD specifically. In addition, future studies should include polygenic scores for AUD in MRS models to investigate the relative contributions to AUD of genetics and epigenetics. While the performance of the MRS does not allow for an immediate clinical application, the current analysis provides an important basis for translational research and testing the clinical applications of the AUD MRS in independent cohorts.
While some of the identified CpG sites showed blood–brain correlations, little to no overlap was observed with findings from recent EWASs of AUD in postmortem human brain tissue35,36. This could be due to the small sample sizes in the human postmortem EWASs, which ranged from 46 to 119 (refs. 35,36). This highlights the importance of combining datasets to enhance statistical power and facilitate the replication of individual study results. Currently, many of the findings from EWASs of AUD in postmortem human brain tissue have not been replicated16,35,36,54, necessitating similar meta-analytical approaches to identify robust findings for which, in turn, the overlap with peripheral blood markers can be determined.
While we present an extensive DNA methylation meta-analysis of alcohol use disorder, the sample size and resulting statistical power remain a potential limitation. The current MRS was developed as an exploratory approach to enhance prediction in a smaller independent sample. In this model, the MRS variable accounted for 10.48% of the variance in heavy drinking, with a predictive AUC-ROC of 0.657, indicating a modest yet promising level of predictive performance. Although these values are comparable to or slightly better than those reported in other EWAS studies of mental disorders, further improvements in predictive performance are needed before such models can be used clinically. In addition, we cannot exclude confounding factors not accounted for in the present analysis that could have driven the associations observed between AUD and DNA methylation. It is important to note that we controlled for factors that could interact with AUD, such as age and sex, but did not examine them as moderators due to sample-size limitations. Future studies in larger samples are needed to model interaction effects appropriately. Due to the correlational nature of the analysis, it is unclear whether DNA methylation at the identified CpG sites is altered because of heavy drinking associated with AUD or whether the DNA methylation itself contributes to the risk of developing AUD. Another limitation is the analysis of bulk tissue that can mask cell type-specific effects of DNA methylation. While we controlled for the major cell type proportions in whole blood, future studies on the single-cell level or using sorted cell populations should be performed to directly investigate cell type-specific DNA methylation signatures of AUD. While we can make assumptions about how differential DNA methylation could influence biological processes through pathway analyses, future studies should also investigate how DNA methylation signatures of AUD affect other -omics signatures, such as transcriptomics. It has been shown that multi-omics data integration, through network approaches, such as WGCNA55, or factor analytical frameworks, such as Multi-Omics Factor Analysis56, can yield more functional insights into disease mechanisms in AUD57,58 and other substance use disorders59,60,61.
Taken together, we identified differentially methylated CpG sites and regions robustly associated with AUD in more than 3,700 participants across 7 cohorts representing heterogeneous AUD phenotypes. Our results identified new targets and reproduced findings from previous EWASs. The resulting MRS was predictive of heavy drinking in an independent cohort, underscoring the potential for use of the MRS in research and eventually in the clinic.
Methods
Samples
Study protocols of all cohorts that participated in the meta-analysis were approved by the respective ethics committees (Supplementary Text 1). All participants provided informed consent, and all procedures were performed in accordance with the Declaration of Helsinki. DNA methylation data from peripheral blood samples of a total of 3,775 individuals, 1,325 individuals with and 2,450 without AUD, from 7 cohorts, all members of the Substance Use Disorder Epigenetics Working Group of the Psychiatric Genomics Consortium, were included in the meta-analysis. Three cohorts focused explicitly on individuals with AUD: the CIMH cohort (N = 194) followed male patients who underwent withdrawal treatment for AUD and matched controls17; in the UCSF Family Alcoholism Study (N = 442), families with an increased risk for AUD were investigated20; and the NIAAA cohort investigated patients seeking treatment for AUD at the NIAAA (N = 615) (ref. 15). The Yale–Penn cohort (N = 231) (refs. 18,19) included participants recruited because they had substance dependence traits or were controls, and the GTP (N = 650) (ref. 21) focused on veterans with PTSD, which is highly comorbid with substance use disorders, such as AUD. Similarly, the NESDA study (N = 1,132) recruited participants with depression and anxiety25, also highly comorbid with AUD. GSMS (N = 540) (ref. 22) is a longitudinal study from early childhood to adulthood enriched for participants with a higher risk of developing a substance use disorder, though only adult time points are considered here. Additional cohort characteristics are described in Supplementary Information. In all cohorts, AUD was diagnosed according to the DSM-IV (alcohol dependence) or DSM-51 (at least moderate AUD).
DNA methylation
DNA methylation levels were determined in five cohorts using the Illumina Infinium Human Methylation EPIC BeadChip v1.0 (about 850,000 probes) or the 450 K BeadChip (about 450,000 probes). In NESDA and GSMS, MBD-seq was performed.
Statistics and reproducibility
This study included samples that had been characterized for AUDs and for which DNA methylation data were available from the Psychiatric Genomics Consortium Substance Use Disorder Workgroup. No statistical method was used to predetermine sample size.
Quality control of raw data
A standardized quality-control pipeline was distributed among all cohorts with microarray data, including the replication cohort LURIC, a customized version of the CPACOR (control probe adjustment and reduction of global correlation) pipeline published by Lehne et al.62. Raw IDAT files were read and processed using the minfi package (version 1.52.1). Illumina background subtraction was performed to remove outliers by removing the average negative control signal intensity. A principal component analysis of the positive control probe intensities was performed to remove technical biases in the EWAS. Probes were excluded if the detection P value was larger than 10−16, resulting in a marker call rate lower than 95%, if the beadcount was lower than 3 in more than 5% of samples, and if probes were located on the sex chromosomes. Samples were removed if the sample call rate was below 95% or if the reported sex did not match biological sex. Cell type composition was estimated using the EpiDISH package (version 2.22.0)63, resulting in estimates for CD8T cells, CD4T cells, natural killer cells (NK), B cells (B), monocytes (mono) and neutrophils.
The quality-control procedure for MBD-seq is described in detail in Clark et al.16. Briefly, sequence reads were aligned to the human reference genome (hg19/GRCh37) using Bowtie264. Quality control of samples, reads and sites was conducted using RaMWAS65 as described elsewhere25,66 and excluded samples with failed enrichment or if reported sex did not match biological sex, resetting read counts if there were excessive duplicate reads (>3 reads starting at the same location were reset to 1), excluding reads in difficult-to-align loci and excluding sporadically methylated sites or those located on sex chromosomes. Using a non-parametric estimate of fragment size distribution, methylation scores were calculated by estimating the number of fragments covering each CpG. These scores represent a quantitative measure of methylation for each sample at a specific CpG. Cell type composition was estimated using Houseman’s method67 and a reference set of methylomes based on MBD-seq data described elsewhere16, resulting in estimates for CD15 granulocytes, CD3T cells, B cells and monocytes.
Cohort-level EWASs
A standardized analysis plan was distributed among participating cohorts. All cohorts with microarray DNA methylation data were additionally provided with a standardized analysis pipeline, while the MBD-seq cohorts ran similar models based on the analysis plan. Before running the EWASs, all variables were tested for multicollinearity by calculating Pearson correlations and flagging variable pairs with a correlation coefficient greater than 0.70. For the cohort-level EWASs, DNA methylation beta values were predicted by AUD status in robust linear models using the R package MASS (version 7.3-65). All cohorts controlled for age, smoking, cell type proportions (CD8T, CD4T, NK, B, mono), three genotype principal components and four principal components of the internal control probes. Cohort-specific covariates were sex and covariates based on cohort ascertainment, such as MDD, PTSD diagnosis. Detailed descriptions of cohort specificities are provided in Supplementary Text 1.
Quality control and filtering of EWAS summary statistics
We performed additional filtering for cross-reactive probes and single nucleotide polymorphisms in base extensions with a minor allele frequency >1% in European and African American ancestry55. We used the EWAS_QC function of the R package QCEWAS (version 1.2-3)56 to calculate lambda values for the cohort results, inspect the distribution of estimates and standard errors, and explore individual-level results. For the primary meta-analysis, MBD-seq results were restricted to CpG sites on the EPIC BeadChip, and the number of sites included is presented in Table 1.
Meta-analysis
The quality-controlled and filtered CpG sites were utilized for an invariance-weighted fixed-effects meta-analysis using METAL (version 2011-03-25)68. In addition, a complementary random-effects meta-analysis was performed using random-metal69 (https://github.com/explodecomputer/random-metal). The meta-analysis focused on CpG sites in at least N = 1,929 samples or at least 50% of the samples, resulting in 25,160 skipped and 708,385 analyzed CpG sites. In addition to the primary meta-analysis, we performed secondary analyses based on methylation quantification, MBD-seq and microarray. These results were subsequently meta-analyzed, and correlation analysis investigating the convergence between the effect sizes of the primary meta-analysis and the secondary meta-analysis was performed. Results from the main meta-analysis were slightly inflated (λ = 1.28) and were subsequently bacon-corrected (version 1.34)70. All results were corrected for multiple testing using the Bonferroni correction. Results were annotated using the manufacturer’s manifest and visualized using the miamiplot R package71.
Downstream analyses
DMRs
DMRs were identified using the comb-p algorithm72, as implemented in ENmix (version 1.42.2)73. Comb-p can be used solely on summary statistics and is therefore well suited for meta-analysis results. The analysis was performed with a seed P value of <1 × 10−5, which determines the initial selection of DMRs, and a maximum distance between CpG sites of 1,000 base pairs. Results were corrected for multiple testing using the FDR.
Gene ontology overrepresentation analysis
DMPs and DMRs were annotated to genes using the manufacturer’s manifest. Then Gene Ontology Overrepresentation Analysis (GO ORA) was conducted at the gene level using clusterProfiler (version 4.14.6)74. In addition, a semantic clustering analysis was performed to identify clusters of related GO terms.
GWAS enrichment analysis
For GWAS enrichment analysis, we formed three gene sets. The first included all genes associated with DMPs (81 genes), the second reduced these CpG sites to those known to be influenced by genotype26 (45 genes), and the third included those associated with the DMRs (81 genes). We then utilized MAGMA (Multi-marker Analysis of GenoMic Annotation, version 1.10)75 to test the enrichment of these gene sets in the summary statistics of a recent trans-ancestry meta-analysis of problematic alcohol use and AUD in more than one million participants27. To investigate whether genetic variation in smoking behavior-associated genes was driving the results, we also performed a GWAS enrichment analysis for a GWAS of cigarettes per day30. In addition, we investigated the association with comorbid disorders, such as MDD31 and PTSD32.
Overlap with alcohol consumption EWAS
To determine whether the epigenome-wide significant CpG sites identified in the primary analysis were influenced by alcohol consumption, we examined the overlap with the findings of a recent EWAS of alcohol consumption involving more than 8,000 individuals from the Generation Scotland cohort33. Fisher’s exact test was used to determine the statistical significance of the overlap.
Blood–brain concordance
We further investigated the blood–brain concordance of CpG sites identified in the primary meta-analysis. In the first step, we used BECon34 to determine the blood–brain correlations of the 118 identified CpG sites. In addition, we performed a ‘look-up’ of suggestive significant CpG sites (P < 1 × 10−5)76 in two recent EWASs of AUD in the dorsolateral prefrontal cortex/Brodmann area35,36, ventral striatum/nucleus accumbens35,36, caudate nucleus35, putamen35, and anterior cingulate cortex35.
Sensitivity analysis for smoking
In some cohorts, a substantial overlap between smoking and AUD was observed (for example, CIMH). Therefore, we performed sensitivity analyses separately for currently self-reported non-smoking participants in all cohorts with at least 30 individuals for AUD cases and 30 for controls. The non-smoking meta-analysis consisted of 939 individuals from four cohorts: NIAAA (N = 369), UCSF (N = 108), NESDA (N = 377) and GSMS (N = 85). Effect sizes from this meta-analysis were associated with those of the primary meta-analysis using Pearson correlation.
Leave-one-out analysis
We further investigated heterogeneity across studies by performing leave-one-out analyses, in which we performed the main meta-analysis seven times, excluding one cohort at a time.
Validation cohort
DNA methylation data from the LURIC was used to replicate MRSs from the meta-analysis in an independent cohort. The LURIC study comprises 3,316 individuals hospitalized for coronary angiography between 1997 and 200037. DNA methylation was assayed using the Illumina EPIC BeadChip v1, and the same quality-control procedures for array-based data in the discovery analysis were applied; details can be found in Supplementary Text 2. After quality control, data were available for 2,534 individuals. All data presented in this paper refer to the subsample with DNA methylation data.
The mean age in the LURIC cohort was M = 62.85 years (s.d. = 10.6), and 30.7% of participants were female (N = 780). Alcohol consumption was assessed by self-report and converted into grams of ethanol per day66. The mean alcohol consumption in grams per day was M = 16.61, s.d. = 24.46, while the median ethanol intake per day was Md = 3.08. Overall, the LURIC cohort is characterized by a high percentage of heavy drinkers, with 939 (37.05%) meeting heavy-drinking criteria as proposed by NIAAA39. For male participants, the heavy-drinking threshold is drinking more than five drinks per day (70 g) or more than 210 g per week. Female participants fulfill heavy-drinking criteria if they consume more than four drinks per day (56 g) or more than 112 g per week. Heavy drinking is considered a risk factor for AUD, which was not assessed in the LURIC cohort.
MRS
We calculated MRSs using the pruning and thresholding approach suggested by Chen et al.40. Here regional associations between the methylation values of the validation cohort were identified using the CoMeBack algorithm (version 0.1.0)77. During the construction of the MRS, one CpG site per region is included in the score, similar to clumping in polygenic risk scores. The minimum P value and resulting P-value thresholds were constructed from the summary statistics of the meta-analysis, resulting in a minimum P value of 5.12 × 10−17, representing the P value of the strongest association in the primary meta-analysis. We tested 16 thresholds in total, ranging from 0.05 to 5 × 10−17 in increments of 1 decimal point.
To test whether the MRS of AUD was predictive of alcohol consumption and heavy drinking in the LURIC cohort, we constructed a linear model predicting the ethanol intake per day by the MRS while controlling for sex, age and cell type composition, as described for the primary meta-analysis. All numerical covariates were scaled to the mean. In addition, we tested whether the AUD MRS was predictive of heavy drinking, according to NIAAA criteria, in a generalized linear model using the same covariates as described above. Here we calculated Nagelkerkes R2 to determine the goodness of fit, as well as the AUC-ROC, using the R package pROC (v. 1.18.5)67.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Full summary statistics are available via figshare at https://doi.org/10.6084/m9.figshare.30000022 (ref. 78). Access to raw DNA methylation data or other individual-level data can be requested for each individual cohort.
Code availability
The code for analysis and visualization is available via GitHub at https://github.com/lzillich/DNAm_AUD_meta and Zenodo at https://doi.org/10.5281/zenodo.19690307 (ref. 79).
References
-
Diagnostic and Statistical Manual of Mental Disorders 5th edn (American Psychiatric Association, 2013).
-
Savage, J. E. et al. Genetic heterogeneity across dimensions of alcohol use behaviors. Am. J. Psychiatry 181, 1006–1017 (2024).
-
Stone, A. L., Becker, L. G., Huber, A. M. & Catalano, R. F. Review of risk and protective factors of substance use and problem use in emerging adulthood. Addict. Behav. 37, 747–775 (2012).
-
Global Status Report on Alcohol and Health 2018 (World Health Organization, 2019).
-
Lohoff, F. W. Targeting unmet clinical needs in the treatment of alcohol use disorder. Front. Psychiatry 13, 767506 (2022).
-
Longley, M. J., Lee, J., Jung, J. & Lohoff, F. W. Epigenetics of alcohol use disorder—a review of recent advances in DNA methylation profiling. Addict. Biol. 26, e13006 (2021).
-
Wedemeyer, F. et al. Prospects of genetics and epigenetics of alcohol use disorder. Curr. Addict. Rep. 7, 446–452 (2020).
-
Gibney, E. R. & Nolan, C. M. Epigenetics and gene expression. Heredity 105, 4–13 (2010).
-
Ducci, F. & Goldman, D. The genetic basis of addictive disorders. Psychiatr. Clin. North Am. 35, 495–519 (2012).
-
González-Reimers, E. et al. Alcoholism: a systemic proinflammatory condition. World J. Gastroenterol. 20, 14660–14671 (2014).
-
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
-
Liu, C. et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 23, 422–433 (2018).
-
Bollepalli, S., Korhonen, T., Kaprio, J., Anders, S. & Ollikainen, M. EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics 11, 1469–1486 (2019).
-
Clark, S. L. et al. Next-generation biomarkers for alcohol consumption and alcohol use disorder diagnosis, prognosis, and treatment: a critical review. Alcohol. Clin. Exp. Res. 49, 5–24 (2025).
-
Lohoff, F. W. et al. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol. Psychiatry 26, 2224–2237 (2021).
-
Clark, S. L. et al. Dual methylation and hydroxymethylation study of alcohol use disorder. Addict. Biol. 27, e13114 (2022).
-
Witt, S. H. et al. Acute alcohol withdrawal and recovery in men lead to profound changes in DNA methylation profiles: a longitudinal clinical study. Addiction 115, 2034–2044 (2020).
-
Pathak, G. A. et al. Epigenetic and genetic profiling of comorbidity patterns among substance dependence diagnoses. Mol. Psychiatry 30, 4435–4443 (2025).
-
Gelernter, J. et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol. Psychiatry 76, 66–74 (2014).
-
Vieten, C., Seaton, K. L., Feiler, H. S. & Wilhelmsen, K. C. The University of California, San Francisco Family Alcoholism Study. I. Design, methods, and demographics. Alcohol Clin. Exp. Res. 28, 1509–1516 (2004).
-
Gillespie, C. F. et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen. Hosp. Psychiatry 31, 505–514 (2009).
-
Costello, E. J. et al. The Great Smoky Mountains Study of Youth. Goals, design, methods, and the prevalence of DSM-III-R disorders. Arch. Gen. Psychiatry 53, 1129–1136 (1996).
-
van den Oord, C. et al. DNA methylation signatures of childhood trauma predict psychiatric disorders and other adverse outcomes 17 years after exposure. Mol. Psychiatry 27, 3367–3373 (2022).
-
Penninx, B. W. et al. The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int. J. Methods Psychiatr. Res. 17, 121–140 (2008).
-
Aberg, K. A. et al. Methylome-wide association findings for major depressive disorder overlap in blood and brain and replicate in independent brain samples. Mol. Psychiatry 25, 1344–1354 (2020).
-
Villicaña, S. et al. Genetic impacts on DNA methylation help elucidate regulatory genomic processes. Genome Biol. 24, 176 (2023).
-
Zhou, H. et al. Multi-ancestry study of the genetics of problematic alcohol use in over 1 million individuals. Nat. Med. 29, 3184–3192 (2023).
-
Walters, R. K. et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 21, 1656–1669 (2018).
-
Saunders, G. R. B. et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature 612, 720–724 (2022).
-
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
-
Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Trans-ancestry genome-wide study of depression identifies 697 associations implicating cell types and pharmacotherapies. Cell 188, 640–652.e9 (2025).
-
Nievergelt, C. M. et al. Genome-wide association analyses identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder. Nat. Genet. 56, 792–808 (2024).
-
Lohoff, F. W. et al. Epigenome-wide association study of alcohol consumption in N = 8161 individuals and relevance to alcohol use disorder pathophysiology: identification of the cystine/glutamate transporter SLC7A11 as a top target. Mol. Psychiatry 27, 1754–1764 (2022).
-
Edgar, R. D., Jones, M. J., Meaney, M. J., Turecki, G. & Kobor, M. S. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 7, e1187 (2017).
-
Zillich, L. et al. Epigenome-wide association study of alcohol use disorder in five brain regions. Neuropsychopharmacology 47, 832–839 (2022).
-
White, J. D. et al. Alcohol use disorder-associated DNA methylation in the nucleus accumbens and dorsolateral prefrontal cortex. Biol. Psychiatry Glob. Open Sci. 4, 100375 (2024).
-
Winkelmann, B. R. et al. Rationale and design of the LURIC study—a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics 2, S1–S73 (2001).
-
Moissl, A. P. et al. Alcohol consumption and mortality: the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Atherosclerosis 335, 119–125 (2021).
-
Understanding alcohol drinking patterns. NIAAA https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-drinking-patterns (2025).
-
Chen, J. et al. Pruning and thresholding approach for methylation risk scores in multi-ancestry populations. Epigenetics 18, 2187172 (2023).
-
Hillary, R. F. et al. Blood-based epigenome-wide analyses of 19 common disease states: a longitudinal, population-based linked cohort study of 18,413 Scottish individuals. PLoS Med. 20, e1004247 (2023).
-
Christiansen, C. et al. Novel DNA methylation signatures of tobacco smoking with trans-ethnic effects. Clin. Epigenetics 13, 36 (2021).
-
Leong, C. et al. Association of alcohol use disorder on alcohol-related cancers, diabetes, ischemic heart disease and death: a population-based, matched cohort study. Addiction 117, 368–381 (2022).
-
Vancampfort, D. et al. The prevalence of diabetes mellitus type 2 in people with alcohol use disorders: a systematic review and large scale meta-analysis. Psychiatry Res. 246, 394–400 (2016).
-
Sung, C. et al. Risk of cardiovascular disease in patients with alcohol use disorder: a population-based retrospective cohort study. PLoS ONE 17, e0276690 (2022).
-
Maleki, N., Tahaney, K., Thompson, B. L. & Oscar-Berman, M. At the intersection of alcohol use disorder and chronic pain. Neuropsychology 33, 795–807 (2019).
-
Stelzer, G. et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics 54, 1.30.1–1.30.33 (2016).
-
McClintick, J. N. et al. Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 47, 505–515 (2013).
-
Liu, Y. et al. Autophagy alleviates ethanol-induced memory impairment in association with anti-apoptotic and anti-inflammatory pathways. Brain Behav. Immun. 82, 63–75 (2019).
-
Scalise, M. et al. Crossing the borders: the amino acid transporter LAT1 (SLC7A5) in the blood–brain barrier. Neurochem. Int. 191, 106070 (2025).
-
Wei, J. et al. Blood–brain barrier integrity is the primary target of alcohol abuse. Chem. Biol. Interact. 337, 109400 (2021).
-
Pla, A., Pascual, M. & Guerri, C. Autophagy constitutes a protective mechanism against ethanol toxicity in mouse astrocytes and neurons. PLoS ONE 11, e0153097 (2016).
-
Shen, X. et al. A methylome-wide association study of major depression with out-of-sample case-control classification and trans-ancestry comparison. Nat. Ment. Health 3, 1152–1167 (2025).
-
Andrade-Brito, D. E. et al. Neuronal-specific methylome and hydroxymethylome analysis reveal significant loci associated with alcohol use disorder. Front. Genet. 15, 1345410 (2024).
-
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
-
Argelaguet, R. et al. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 14, e8124 (2018).
-
Zillich, L. et al. Multi-omics signatures of alcohol use disorder in the dorsal and ventral striatum. Transl. Psychiatry 12, 190 (2022).
-
Zillich, L. et al. Multi-omics analysis of alcohol effects on the liver in young and aged mice. Addict. Biol. 28, e13342 (2023).
-
Zillich, E. et al. Multi-omics profiling of DNA methylation and gene expression alterations in human cocaine use disorder. Transl. Psychiatry 14, 428 (2024).
-
Zillich, E. et al. A multi-omics and cell type-specific characterization of the ventral striatum in human cocaine use disorder. Cell Rep. 44, 115332 (2025).
-
Zillich, L. et al. Cell type-specific multi-omics analysis of cocaine use disorder in the human caudate nucleus. Nat. Commun. 16, 3381 (2025).
-
Lehne, B. et al. Erratum to: A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 17, 73 (2016).
-
Zheng, S. C., Breeze, C. E., Beck, S. & Teschendorff, A. E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 15, 1059–1066 (2018).
-
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
-
Shabalin, A. A. et al. RaMWAS: fast methylome-wide association study pipeline for enrichment platforms. Bioinformatics 34, 2283–2285 (2018).
-
Clark, S. L. et al. Methylomic investigation of problematic adolescent cannabis use and its negative mental health consequences. J. Am. Acad. Child Adolesc. Psychiatry 60, 1524–1532 (2021).
-
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
-
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
-
Hemani, G. explodecomputer/random-metal: adding random effects model. Zenodo https://doi.org/10.5281/zenodo.6974696 (2022).
-
van Iterson, M., van Zwet, E. W. & Heijmans, B. T. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 18, 19 (2017).
-
White, J. D. MiamiPlot: an R package for creating ggplot2 based miami plots. GitHub https://github.com/juliedwhite/miamiplot (2020).
-
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
-
Xu, Z., Niu, L. & Taylor, J. A. The ENmix DNA methylation analysis pipeline for Illumina BeadChip and comparisons with seven other preprocessing pipelines. Clin. Epigenetics 13, 216 (2021).
-
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
-
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
-
Schiele, M. A. et al. Epigenome-wide DNA methylation in unipolar depression: predictive biomarker of antidepressant treatment response? Int. J. Neuropsychopharmacol. 27, pyae045 (2024).
-
Gatev, E., Gladish, N., Mostafavi, S. & Kobor, M. S. CoMeBack: DNA methylation array data analysis for co-methylated regions. Bioinformatics 36, 2675–2683 (2020).
-
Zillich, L. et al. Full summary statistics for ‘A large-scale meta-analysis of DNA methylation signatures of alcohol-use disorder in the Psychiatric Genomics Consortium’. figshare https://doi.org/10.6084/m9.figshare.30000022 (2026).
-
Zillich, L. lzillich/DNAm_AUD_meta: manuscript_publication_v1.0. Zenodo https://doi.org/10.5281/zenodo.19690307 (2026).
Acknowledgements
We thank all participants in the studies supporting this report.
Funding
L.Z. discloses support for the research of this work from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [551108212]. F.W.L. discloses support for publication of this work from the Intramural Research Program of the National Institutes of Health (NIH) as part of the Division of Intramural Clinical and Biological Research of the National Institute on Alcohol Abuse and Alcoholism (NIAAA) [ZIA-AA000242]. S.H.W. discloses support for this work from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [402170461] and through the German Federal Ministry of Education and Research (BMBF) through the e:Med consortium “A systems-medicine approach towards distinct and shared resilience and pathological mechanisms of substance use disorders – SysMedSUDs” [01ZX01909]. R.S. discloses support for this publication from the German Federal Ministry of Education and Research (BMBF) through the e:Med consortium “A systems-medicine approach towards distinct and shared resilience and pathological mechanisms of substance use disorders – SysMedSUDs” [01ZX01909].Open access funding provided by Albert-Ludwigs-Universität Freiburg im Breisgau.
Ethics declarations
Competing interests
H.R.K. is a member of advisory boards for Altimmune and Clearmind Medicine; a consultant to Sobrera Pharmaceuticals, Altimmune, and Lilly; the recipient of research funding and medication supplies for an investigator-initiated study from Alkermes and a company-initiated study by Altimmune; and an inventor on US provisional patent “Multi-ancestry Genome-wide Association Meta-analysis of Buprenorphine Treatment Response.” J.M.O. is a current employee and stockholder of Regeneron Pharmaceuticals. J.M.O. contributed to the paper as an analyst while affiliated with the University of Missouri. M.E.K. and W.M. are employees of Synlab Holding Deutschland GmbH. K.D. has been a member of the Janssen-Cilag GmbH Steering Committee Neurosciences and received speaker’s honoraria from Janssen-Cilag GmbH until 2022. Currently, K.D. is a member of the Neurotorium Editorial Board, Lundbeck Foundation. The other authors declare no competing interests.
Peer review
Peer review information
Nature Mental Health thanks Charlotte Cecil, Mitsuru Kimura and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quantile-quantile plots of meta-analyses and cohort-level EWASs.
Quantile-quantile (QQ) plots depicting expected versus observed p values of the A) bacon-corrected and B) uncorrected meta-analysis results and cohort-level EWAS linear models for C) Central Institute of Mental Health (CIMH) cohort, D) Great Smoky Mountain Study (GSMS), E) Grady Trauma Project (GTP), F) National Institute on Alcohol Abuse and Alcoholism (NIAAA), G) University of California at San Francisco Family Alcoholism Study (UCSF), H) Netherlands Study of Depression and Anxiety (NESDA), I) Yale-Penn cohort.
Extended Data Fig. 2 Overlap of effect sizes of array- and MBD-seq-based meta-analysis and differentially methylated regions.
A)-B): scatterplot depicting the correlation between effect sizes of the array-only meta-analysis with the effect sizes of the main analysis for A) all CpG sites significantly associated with alcohol use disorder (Bonferroni- and Bacon-corrected p-value < 0.05) and B) CpG sites nominally significant in the main analysis (uncorrected p-value < 0.05). All tests have been performed two-sided. C) Manhattan plot depicting differentially methylated regions (DMRs); genes of the twelve most significant DMRs are highlighted; blue dashed line indicates epigenome-wide significance. All tests have been performed two-sided.
Extended Data Fig. 3 Gene Ontology overrepresentation analysis.
Visualization of Gene Ontology overrepresentation analysis (GO ORA) results. A) overview of most significant GO ORA results for differentially methylated CpG sites; and B) DMRs; circle size represents gene counts, pink dots indicate molecular functions (MF), green dots cellular components (CC), and blue dots biological processes (BP). All tests have been performed two-sided.
Extended Data Fig. 4 Blood-brain concordance.
Correlation between blood and brain DNA methylation for CpG sites associated with AUD in primary meta-analysis. BA = Brodmann Area.
Extended Data Fig. 5 Smoking sensitivity analysis.
Scatterplots of effect sizes of meta-analysis in individuals who did not report to smoke and the primary meta-analysis for all CpG sites tested (upper panel) and CpG sites nominally significant in the primary meta-analysis (lower panel, uncorrected p-value < 0.05).
Extended Data Fig. 6 Results of leave-one-out analyses.
Results of the leave-one-out analyses. Regression coefficients for AUD for the six most significant CpG sites in the meta-analysis with converging effect estimates in at least five cohorts; dots represent unstandardized coefficients from the meta-analyses, error bars represent standard errors of estimate, diamond shape represents effect in meta-analysis; Each line depicts the regression coefficients for the leave-one-out analysis excluding: YP_out = Yale-Penn (N = 3,544), UCSF_out = University of California at San Francisco (N = 3,333), NIAAA_out = National Institute on Alcohol Abuse and Alcoholism (N = 3,160), NESDA_out = Netherlands Study of Depression and Anxiety (N = 2,643), GTP_out = Grady Trauma Project (3,098), GSMS_out = Great Smoky Mountain Study (N = 3,291), CIMH_out = Central Institute of Mental Health (N = 3,581). META = primary meta-analysis (N = 3,775).
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zillich, L., D’Augello, S., Avetyan, D. et al. A large-scale meta-analysis of DNA methylation signatures of alcohol use disorder in the Psychiatric Genomics Consortium. Nat. Mental Health (2026). https://doi.org/10.1038/s44220-026-00666-w
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s44220-026-00666-w
Leave a Reply