
⏱ 11 min read
Schizophrenia’s genetic architecture sits awkwardly between neurons and the immune system. A 2026 CRISPR screen of 30 schizophrenia-associated genes in human microglia-like cells found that several risk genes meaningfully alter how microglia engulf synaptic material, supporting the synaptic-pruning model at the functional gene level.1
Research Highlights
- 30 schizophrenia risk genes were knocked out one by one in human microglia-like cells. Horng et al. used arrayed CRISPR (Cas9 ribonucleoproteins delivered to PBMC-derived microglia-like cells, or piMGLCs) on genes prioritized from postmortem dorsolateral prefrontal cortex transcriptomes plus phagocytosis-related ontology lists.1
- Most knockouts reduced synaptic engulfment; 7 increased it. ITGB2 and ITGAM (CR3 complement-receptor subunits) targeting raised phagocytic index. MSR1 and CYFIP1 targeting reduced it. Effects replicated across primary and secondary screens with redesigned guide RNAs (R² = 0.94 excluding 1 outlier).1
- Morphology tracked phagocytosis. Ameboid microglia (round, less branched) were more phagocytic; ramified microglia (highly branched, surveillance-state) less so. Solidity vs. phagocytic index R² = 0.56; CD68 activation marker vs. phagocytic index R² = 0.85.1
- TREM2, SYK, and IRF8 reshaped the broader transcriptome. Knocking out these 3 genes drove the largest number of differentially expressed downstream genes, hitting phagocytosis, lysosomal acidification, antigen presentation, and inflammatory signaling pathways simultaneously.1
- This strengthens but does not prove the pruning hypothesis. The screen is in vitro, in microglia-like cells from healthy donors, with a 6-day editing window. It supports a molecular pruning role for these risk genes; it does not show that human schizophrenia is caused by these effects.
The pruning hypothesis of schizophrenia traces back to Feinberg’s 1982 observation that schizophrenia onset overlaps with adolescent synapse elimination, and to postmortem studies showing reduced dendritic spines in schizophrenia cortex.2 Sekar et al. 2016 then connected the strongest schizophrenia risk locus — structural variants in the C4 complement gene — to higher C4A expression and, in mouse models, increased complement-mediated synaptic pruning by microglia.3
What Horng et al. add is mechanism at the gene level. Most schizophrenia genome-wide association study (GWAS) hits are not C4.
The 2022 Trubetskoy et al. mega-analysis of 320 schizophrenia loci showed convergence on synaptic biology, but also flagged immune-related genes whose function in microglia was unclear.4 A CRISPR screen tests, gene by gene, whether disrupting those candidates actually changes microglial behavior in a human cell context.
The Screen: 30 Risk Genes, Arrayed CRISPR, Synaptosome Phagocytosis Readout
The system is worth understanding. piMGLCs (PBMC-induced microglia-like cells) are made by taking peripheral blood mononuclear cells — ordinary white blood cells — and treating them with IL-34 plus GM-CSF for 8 days, which transdifferentiates them into cells that express microglial markers and behave like microglia in functional assays. They are not perfect surrogates for brain microglia, but they let researchers screen primary human cells from many donors at scale.
The 30 target genes were chosen by intersecting 3 lists: schizophrenia-associated postmortem differentially expressed genes from the dorsolateral prefrontal cortex, genes expressed in human microglia, and genes flagged by KEGG/GSEA ontology analyses as involved in phagocytosis pathways.
From a starting pool of around 200 candidates, 30 made the cut.1
The primary readout was phagocytic index. Researchers fed the cells synaptosomes (purified synaptic terminals from human iPSC-derived neurons, labeled with a pH-sensitive red fluorophore that lights up only in acidic phagolysosomes), then quantified engulfment by high-content confocal imaging.
Each gene was knocked out using 3 guide RNAs delivered as Cas9-RNP complexes to 25,000 cells per well, with non-targeting guides as controls. Top hits then went into a secondary screen with redesigned guides plus an extra eGFP non-targeting control.
The strongest validation criterion is replication across screens with independent sgRNAs — if 2 different guides hitting different parts of the same gene produce the same phagocytic phenotype, the effect is unlikely to be off-target. Across genes (excluding ITGAM, which behaved as a clear outlier between screens), primary-vs-secondary correlation was R² = 0.94.1
ITGB2, ITGAM, MSR1, and CYFIP1 Are the Strongest Phagocytosis Hits
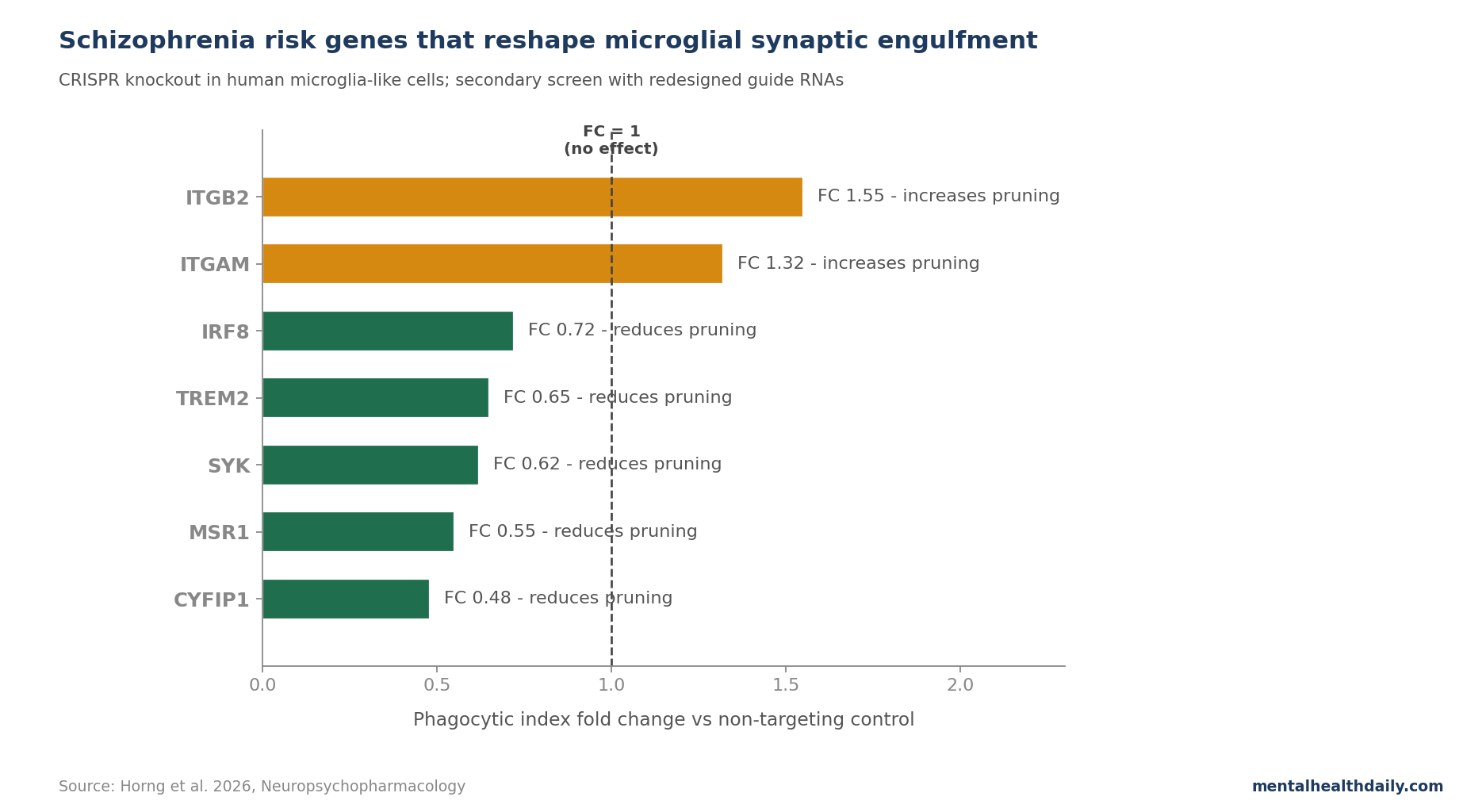
ITGB2 and ITGAM encode the 2 subunits of complement receptor 3 (CR3), the best-characterized microglial receptor for tagging and engulfing complement-coated synapses. Knocking either out raised phagocytic index in this assay — a counterintuitive direction, since CR3 is supposed to drive phagocytosis.
Possible explanations include cellular state shifts (the cells become more ameboid and broadly phagocytic by other receptor pathways) or a non-CR3 regulatory role for these subunits in resting microglia. The 2 genes also produced strikingly different transcriptional fingerprints — ITGB2 knockout had roughly 5 times as many differentially expressed downstream genes as ITGAM, which argues against simple redundancy.1
MSR1 (macrophage scavenger receptor 1) and CYFIP1 (cytoplasmic FMR1-interacting protein 1) targeting reduced phagocytic index. CYFIP1 sits on chromosome 15q11.2 in a copy-number-variable region linked to schizophrenia, autism, and intellectual disability; the gene contributes to actin polymerization through the WAVE complex, which is plausibly required for the cytoskeletal changes that engulfment demands.
None of the gene knockouts reduced cell viability vs. controls, which matters: a phagocytosis “deficit” that is just dead cells is not a phagocytosis deficit. Live-cell imaging confirmed cells stayed viable across the 6-day post-editing window.1
Morphology Tracks Function: Ameboid Microglia Eat More
A long-standing question is whether the morphological “activation” states neuropathologists describe in postmortem schizophrenia brain — ramified surveillance-state microglia vs. ameboid activated states — correspond to functional differences in pruning. The screen offers a direct test.
Solidity (how filled-in the cell shape is — high = round/ameboid, low = branched/ramified) correlated positively with phagocytic index (R² = 0.56). Eccentricity (how elongated the cell is) correlated negatively (R² = 0.48). And CD68 expression, a canonical lysosomal activation marker, correlated tightly with phagocytic index (R² = 0.85).1
Translation: when these knockouts make the cells look activated under a microscope, they tend to actually be activated functionally — eating more synaptic material with more lysosomal machinery turned on. The morphology-state shorthand is biologically meaningful, at least in this model.
This is consistent with patient-derived data. Sellgren et al. 2019 showed that microglia-like cells derived from patients with schizophrenia engulfed more synaptic material than cells from controls, with the difference reversed by minocycline.5 The Horng screen suggests which specific risk genes might mediate that excess.
TREM2, SYK, and IRF8 Are Broad Regulatory Hubs
The transcriptomic arm of the study (DRUG-seq across 30 knockouts vs. controls, 3 biological replicates each) ranks genes by how widely each one perturbs the rest of the microglial transcriptome. TREM2, IRF8, HLA-DMB, and SYK caused the most widespread differential expression, while ITGB2, ITGAM, NCKAP1L, and TLR2 had narrower, more pathway-focused effects.
TREM2 (triggering receptor expressed on myeloid cells 2) is best known as an Alzheimer’s risk gene; the screen shows it also reshapes microglial state in schizophrenia-relevant directions, downregulating CCL3, SPHK1, and CLU while upregulating Fc gamma receptors and cathepsin C.
SYK (spleen tyrosine kinase) is a downstream signaling hub for TREM2, Fc receptors, and ITGB2-coupled CR3. The transcriptional response to SYK knockout overlapped strongly with the TREM2 response, supporting their shared pathway.
IRF8 (interferon regulatory factor 8) is a transcription factor that controls broad swathes of microglial identity. Knocking it out increased pro-inflammatory transcripts — an activation response — even as phagocytosis dropped. The dissociation is biologically useful: IRF8 affects engulfment and shifts the global microglial program.1
23% of the gene-pair interactions Horng et al. identified matched documented STRING-database interactions; 41% were novel and may represent microglia-specific regulatory networks not yet annotated in protein-interaction databases.
Microglial Gene Hits Strengthen the Schizophrenia Pruning Hypothesis
The pruning hypothesis is roughly: in healthy adolescence, microglia eliminate redundant or weakly active synapses to refine cortical circuits. In schizophrenia, that elimination runs hot — with too many or the wrong synapses pruned, particularly in prefrontal cortex — and the resulting circuit-level fragility predisposes to psychosis in late adolescence/early adulthood.2
Evidence for the hypothesis rests on 4 pillars:
- Postmortem dendritic-spine reductions in schizophrenia DLPFC layer 3, dating to Glantz & Lewis 2000 and replicated in multiple cohorts.6
- Sekar et al. 2016: a copy-number variant raising C4A complement expression is the single largest schizophrenia risk locus and increases pruning in mice.3
- Sellgren et al. 2019: patient-derived microglia engulf more synaptic material in vitro; minocycline reduces it, and minocycline use in early life was associated with lower schizophrenia incidence in a Finnish registry.5
- Trubetskoy et al. 2022 GWAS: schizophrenia-associated loci enrich for synaptic and immune pathways, including microglial-expressed genes.4
What was missing is functional validation at the individual-gene level. The Horng screen fills part of that gap by showing that several non-C4 schizophrenia risk genes — including ones in the GWAS but not previously functionally tested in microglia — do alter phagocytosis when disrupted.
Limitations and What This Screen Cannot Show
It is in vitro, in a surrogate cell type. piMGLCs are PBMC-derived microglia-like cells, not actual brain microglia. They share key markers and functional behaviors but live in a 96-well plate, not a brain. Brain microglia interact with neurons, astrocytes, blood vessels, and cerebrospinal fluid in ways no plate model captures.
It is a knockout screen, not a risk-allele screen. Disrupting a gene completely is not the same as carrying a small-effect schizophrenia risk variant that subtly tunes its expression. Most GWAS variants change risk by 5–20% of an odds ratio, not by ablation. Whether the same direction of effect holds at the variant level is a separate question.
The donors were healthy controls. The piMGLCs were derived from healthy PBMCs, then perturbed. The screen does not compare schizophrenia patient-derived to control-derived microglia.
Phagocytic index is one phenotype, not synaptic pruning. The assay measures engulfment of purified synaptosomes in a dish over 3 hours. Real synaptic pruning involves spatial recognition of weak synapses, complement tagging, microglial process motility, and developmental timing — none of which a synaptosome-feeding assay captures.
CRISPR knockout efficiency varied across targets. 6 of 30 genes did not show significant transcript-level changes after editing, suggesting some guides failed to cause nonsense-mediated decay even when phagocytic phenotypes appeared. This complicates interpretation for those targets.
SYK, TREM2, and Microglial State Become Drug-Development Targets
The screen reframes which microglial pathways might be drug-tractable. SYK, in particular, is interesting: it’s a kinase, kinases are druggable, and SYK inhibitors are already in clinical use for autoimmune indications (fostamatinib for immune thrombocytopenia).
Whether brain-penetrant SYK inhibition could shift microglial pruning in a clinically useful direction is speculative but no longer purely theoretical.
TREM2 is being pursued as a therapeutic target in Alzheimer’s disease, where most groups are trying to increase TREM2 signaling to enhance amyloid clearance. The schizophrenia direction would likely be the opposite — if too much pruning is the problem, dampening TREM2-coupled microglial activation could reduce synaptic loss. The same target, opposite direction, depending on disease.
Functional screens like Horng’s supply the gene-level map for drug development, but the disease-specific direction has to come from human disease biology, which this paper doesn’t settle.
Questions About Schizophrenia, Microglia, and Synaptic Pruning
Does this study mean schizophrenia is an immune disease?
It supports the long-running idea that microglia — the brain’s resident immune cells — participate in schizophrenia biology, but does not show that classical inflammation is the cause. Microglia have many roles besides inflammation, including synaptic pruning, neurotrophic support, and circuit refinement.4 A microglia-centric model is not the same as an immune-system-attack model.
Could anti-inflammatory drugs treat schizophrenia?
Trials of broad anti-inflammatories (NSAIDs, minocycline, omega-3s) in schizophrenia have produced small and inconsistent effects on positive and negative symptoms, with the strongest signals in early-stage or first-episode populations. Cho et al. 2019 meta-analyzed 14 minocycline trials and found a modest improvement in negative symptoms (SMD around −0.43) but no clear effect on overall outcome.7
The Horng screen suggests broad anti-inflammatory drugs are blunt instruments compared with what a microglia-targeted treatment could be. Specific gene-pathway targeting is the longer game.
Is C4 still the strongest piece of evidence?
Sekar et al. 2016 remains the strongest gene-to-mechanism example in schizophrenia genetics — a structural variant raises C4A expression, more C4A drives more complement-tagged pruning, and the variant carries the largest single risk-locus effect.3
The Horng screen broadens the picture by showing several other risk genes also affect phagocytosis, but does not displace C4’s central role.
Why does CR3 disruption (ITGB2/ITGAM) increase phagocytosis instead of decreasing it?
The simplest explanation is cellular-state compensation: knocking out CR3 subunits shifts microglia toward a more ameboid, broadly activated state that engulfs material through other receptor pathways (Fc receptors, scavenger receptors).1
It also suggests CR3 has regulatory roles beyond “pull tagged synapses in.” The transcriptional fingerprints of ITGB2 vs. ITGAM knockout differed by roughly 5-fold despite the proteins forming a complex, hinting that subunit-specific functions exist beyond their dimer.
Could this lead to a blood test for schizophrenia?
Not directly. The PBMC-derived microglia-like cell platform is research-grade — useful for screening drug candidates and disease-mechanism work, not for individual diagnosis.
A patient-derived version of this assay (comparing piMGLCs from schizophrenia patients vs. controls) is the natural next step and would test whether the in vitro phagocytic phenotype tracks with diagnosis or symptom severity.
References
- Functional genomic profiling of schizophrenia-associated genes reveals key microglial regulators. Horng JE et al. Neuropsychopharmacology. 2026. doi:10.1038/s41386-026-02406-1
- Schizophrenia caused by a fault in programmed synaptic elimination during adolescence? Feinberg I. Journal of Psychiatric Research. 1982;17(4):319-334. doi:10.1016/0022-3956(82)90038-3
- Schizophrenia risk from complex variation of complement component 4. Sekar A et al. Nature. 2016;530(7589):177-183. doi:10.1038/nature16549
- Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Trubetskoy V et al. Nature. 2022;604(7906):502-508. doi:10.1038/s41586-022-04434-5
- Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Sellgren CM et al. Nature Neuroscience. 2019;22(3):374-385. doi:10.1038/s41593-018-0334-7
- Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Glantz LA & Lewis DA. Archives of General Psychiatry. 2000;57(1):65-73. doi:10.1001/archpsyc.57.1.65
- Adjunctive use of anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Cho M et al. Australian & New Zealand Journal of Psychiatry. 2019;53(8):742-759. doi:10.1177/0004867419835028
Leave a Reply