
⏱ 9 min read
A 2026 single-cell and spatial-transcriptomics study of 21,156 brain immune cells found that late amyloid disease shifted interferon-driven neuroinflammation from microglia toward plaque-associated CD8 T cells, with T-cell frequency strongly tracking total amyloid deposits (Spearman rho = 0.844, p < 0.0001).1 The result does not turn Alzheimer’s disease into an autoimmune disease, but it makes the immune timeline less microglia-only: plaques appear to acquire a local adaptive-immune edge as amyloid pathology matures.
Research Highlights
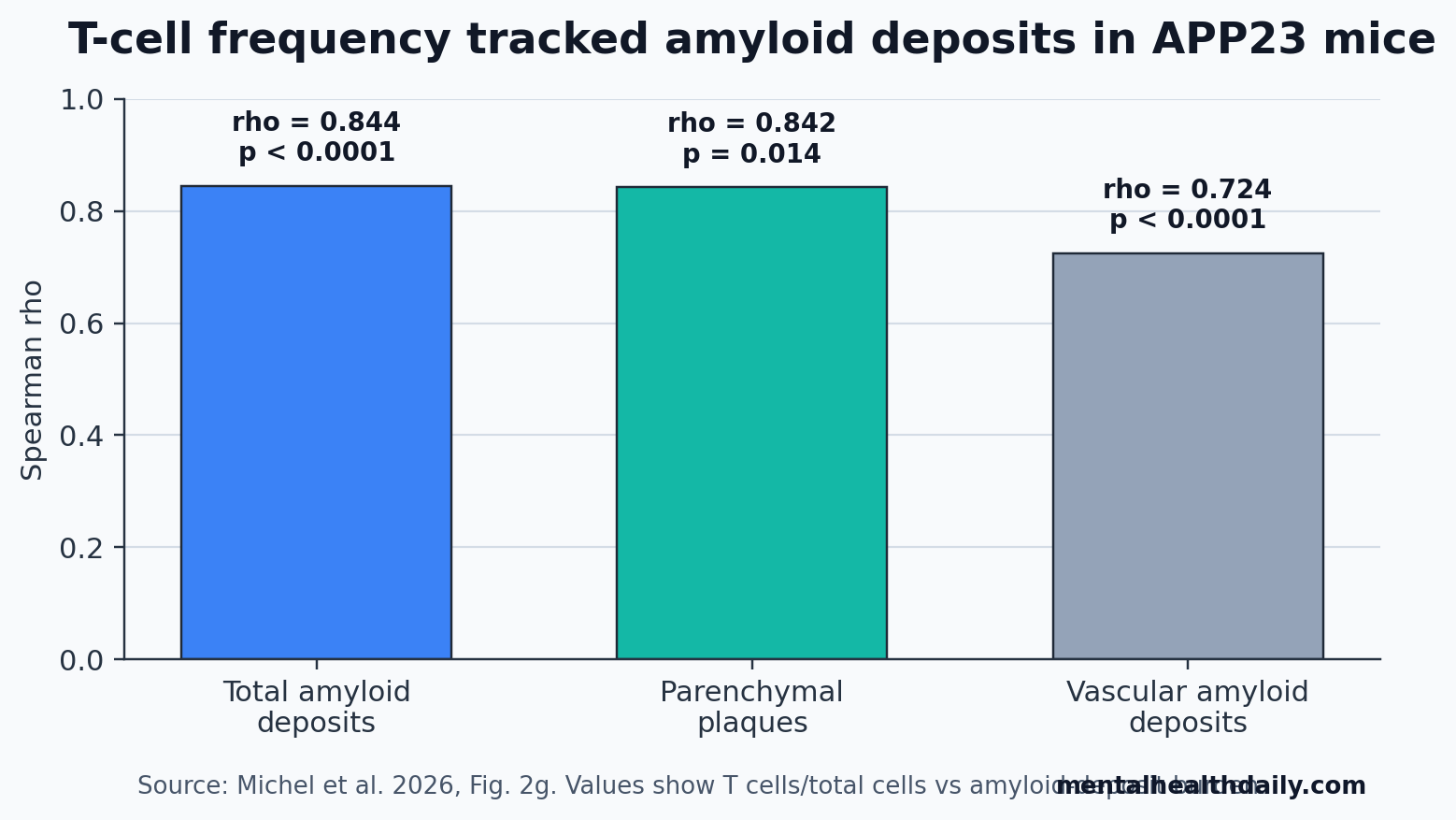
- Late amyloid plaques drew T cells: in APP23 mice, T-cell frequency correlated with total amyloid deposits at rho = 0.844 and with parenchymal plaques at rho = 0.842.
- Interferon timing changed: early amyloid disease was more microglia-centered, while late disease showed a plaque-associated CD8 T-cell subset expressing interferon-stimulated genes.
- CXCL10 was the traffic cue: ISG-positive CD8 T cells produced CXCL10, and CXCR3 knockout or blockade reduced CD8 T-cell migration in transwell assays.
- Human tissue partly matched the mouse pattern: postmortem samples from 4 Alzheimer’s disease patients showed plaque-neighborhood enrichment of interferon-stimulated genes, CXCL10, PDCD1, NKG7, and GZMK.
- Treatment implications remain indirect: the human validation used 4 postmortem Alzheimer’s disease patients, so the paper supports a timing-specific immune mechanism, not immediate T-cell or interferon blockade.
Type I interferon is an antiviral-style inflammatory signaling program that can help cells coordinate immune defense, but chronic activation in brain tissue can also push microglia and T cells into damaging inflammatory states. CD8 T cells are adaptive immune cells best known for killing infected or abnormal cells; in this study, the relevant question was whether amyloid-beta plaques attract and reshape those cells inside the central nervous system.
Amyloid-beta plaques are clumps of amyloid-beta protein in brain tissue and blood-vessel walls. Alzheimer’s research has usually treated microglia, the brain’s resident immune cells, as the first immune responders to plaques. Michel et al. tested whether later amyloid pathology adds another layer: recruited T cells that sit near plaques, adopt an interferon-stimulated state, and draw in more T cells through chemokine signaling.1
Late Amyloid Disease Shifted the Immune Signal From Microglia to CD8 T Cells
The study used APP23 transgenic mice, a model that develops amyloid-beta plaques in brain tissue and cerebral blood vessels. APP23 mice are useful for studying amyloid-driven inflammation because they accumulate amyloid pathology without the tau tangles that complicate full human Alzheimer’s disease.
Researchers sorted CD45-positive immune cells from mouse brain parenchyma and profiled them with single-cell RNA sequencing and T-cell receptor sequencing. The dataset included 21,156 cells and 20 transcriptionally distinct immune-cell clusters across 4 experimental groups: early APP23 mice, late APP23 mice, and age-matched wild-type controls at both timepoints.
Early pattern: microglial pathways were more prominent. Disease-associated microglia and interferon-stimulated microglia fit the familiar amyloid-neuroinflammation model: plaque recognition, phagocytosis, lipid handling, and innate immune activation.
Late pattern: lymphocyte migration, activation, and effector-function pathways became more prominent. CD8 T cells increased in late APP23 disease, while an IL-17-positive CD4 T-cell cluster also rose. The central immune handoff was not “microglia disappear”; it was a shift in the dominant interferon-linked responder near plaques.
T Cells Clustered Around Parenchymal Plaques More Than Vascular Amyloid
Histology put the sequencing result into space. Michel et al. stained amyloid-beta, CD3-positive T cells, and CD31-positive endothelial cells so they could distinguish parenchymal plaques from vascular amyloid deposits. T cells were not evenly scattered through brain tissue. They increased with age, rose with plaque burden, and clustered in plaque-dense regions.
The strongest reader-facing number is the plaque correlation. T-cell frequency tracked total amyloid deposits at rho = 0.844 (p < 0.0001), parenchymal plaques at rho = 0.842 (p = 0.014), and vascular amyloid deposits at rho = 0.724 (p < 0.0001). The parenchymal signal is important because vascular amyloid is more directly exposed to blood-derived immune cells, yet T cells showed a greater pull toward parenchymal plaque neighborhoods.
The researchers defined plaque neighborhoods in the spatial-transcriptomics assay as cells and transcripts inside plaques, adjacent to plaques within 69 μm of the plaque boundary, or outside that radius. Interferon-stimulated genes rose near plaques, especially in late disease, and distance-based analyses pointed more strongly toward T-cell-associated interferon expression than microglia-associated interferon expression in those late plaque neighborhoods.
CXCL10-CXCR3 Signaling Explained How More T Cells Could Be Recruited
CXCL10 is a chemokine, meaning a chemical cue that helps immune cells move toward inflammatory sites. CXCR3 is the receptor that lets T cells respond to CXCL10 and related chemokines. In this paper, the CXCL10-CXCR3 axis turned the finding from a location map into a plausible recruitment mechanism.
Single-cell analyses showed that ISG-positive CD8 T cells were a major CXCL10-producing population in late amyloid disease. Other CD8 and natural-killer-like T-cell populations showed high CXCR3 expression, making them plausible targets for CXCL10-driven recruitment. Spatial transcriptomics then put CXCL10 close to amyloid plaques and close to interferon-stimulated-gene expression.
The functional test mattered. Researchers used CRISPR-Cas9 to knock out CXCR3 in primary splenic T cells, reaching approximately 40% transduction efficiency. In migration assays, recombinant CXCL10 strongly attracted control CD8 T cells, while CXCR3 knockout or CXCR3-blocking antibody reduced migration. Supernatants from interferon-stimulated CD8 T cells also drove migration of control CD8 T cells more than CXCR3-deficient cells.
Mechanistic interpretation: late amyloid plaques may create a local loop in which interferon-stimulated CD8 T cells produce CXCL10, CXCR3-positive T cells move toward that cue, and recruited effector cells expand or exhaust near the plaque edge. The paper also found activation and exhaustion markers near plaques, including Nkg7, Gzmk, Ccl3, Pdcd1, Lag3, and Tox in the mouse analyses.
Human Alzheimer’s Tissue Supported the Plaque-Neighborhood Signal
Mouse amyloid models can overlead if treated as miniature human Alzheimer’s disease. Michel et al. partly addressed that by testing postmortem brain tissue from 4 Alzheimer’s disease patients with targeted spatial single-cell transcriptomics. The human analysis focused on amyloid-plaque neighborhoods, not clinical outcomes.
Interferon-stimulated genes including IFIT1, IFIT2, and IRF7 were enriched inside or near plaques. CXCL10 transcripts were also enriched close to plaques. Activation and exhaustion-associated transcripts, including NKG7, GZMK, and PDCD1, showed plaque-neighborhood patterns consistent with activated or exhausted T cells near amyloid pathology.
Evidence-strength note: the human dataset supports biological plausibility and cross-species spatial overlap. It does not prove that the same T-cell state predicts dementia progression, anti-amyloid-treatment risk, or patient-level response to immune drugs.
Adjacent Alzheimer’s Studies Make the Timing Problem Harder
Gate et al. previously found clonally expanded CD8 T cells in cerebrospinal fluid from patients with mild cognitive impairment or Alzheimer’s disease, suggesting that adaptive immune activity is not confined to mouse models.2 That study made the human T-cell signal visible, but it did not localize the cells around individual amyloid plaques the way the 2026 spatial-transcriptomics analysis did.
Chen et al. pushed the field toward a harmful T-cell model in tauopathy: microglia-mediated T-cell infiltration drove neurodegeneration in tauopathy mice, and T-cell depletion reduced neuronal loss in that setting.3 Jorfi et al. added a human-model bridge by showing that infiltrating CD8-positive T cells worsened Alzheimer’s-like pathology in a 3D human neuroimmune-axis system, increasing microglial activation, neuroinflammation, and neurodegeneration.4
The complication is Marsh et al., which found that adaptive immunity restrained Alzheimer’s-like pathogenesis by modulating microglial function.5 That is not a contradiction to hand-wave away. T-cell effects can depend on disease stage, amyloid vs. tau context, T-cell subset, and whether the experiment removes all T cells early or targets a late inflammatory subset after plaques are established.
The 2026 paper fits best as a timing model: early amyloid inflammation is more microglia-centered, while late plaque neighborhoods can acquire interferon-stimulated CD8 T-cell recruitment. That timing model explains why broad T-cell depletion can look harmful in one design and protective in another.
Why This Does Not Yet Justify Interferon or T-Cell Blockade in Alzheimer’s
Several therapeutic ideas naturally follow from the mechanism: block type I interferon signaling, interrupt CXCL10-CXCR3 recruitment, or target the plaque-associated ISG CD8 T-cell state more specifically. The paper discusses these possibilities because related drugs and pathways already exist in other inflammatory diseases.
The leap from mechanism to treatment is still too large for clinical advice. The necessary missing pieces are straightforward:
- Timing: the same immune pathway may be protective early and damaging late.
- Target specificity: broad T-cell or interferon suppression could impair useful immune surveillance.
- Clinical endpoints: the study did not test memory, cognition, ARIA risk, or treatment response in humans.
- Human staging: postmortem plaque neighborhoods cannot show when the immune switch first appears during living disease.
For anti-amyloid therapy, the result raises a sharper research question: whether rapid plaque removal changes local T-cell recruitment, vessel-adjacent inflammation, or amyloid-related imaging abnormalities. It does not show that patients receiving anti-amyloid antibodies should also receive immune suppression.
Questions About Amyloid Plaques and CD8 T Cells
Did this study show that T cells cause Alzheimer’s disease?
No. It showed that late amyloid pathology in APP23 mice was associated with plaque-neighborhood CD8 T-cell accumulation, interferon-stimulated gene expression, CXCL10 production, and CXCR3-dependent migration. That is a mechanistic inflammatory pathway, not proof that T cells initiate Alzheimer’s disease.
Is this mainly an amyloid paper or an immune paper?
Both, but the article’s useful claim is immune timing. Amyloid plaques are the local organizing lesion; the immune finding is that plaque-associated inflammation appears to move from a more microglia-centered state toward an adaptive CD8 T-cell state as disease advances.
Does this make type I interferon a treatment target?
It makes type I interferon a plausible research target in amyloid neuroinflammation. Treatment claims need longitudinal human staging, cognitive outcomes, safety data, and a way to avoid blocking immune functions that may be protective earlier in disease.
Why did the paper emphasize parenchymal plaques?
T cells tracked parenchymal plaques strongly even though vascular amyloid should be more accessible to blood-derived immune cells. That pattern suggests the local plaque microenvironment, not simple blood exposure, helps shape T-cell recruitment.
References
- Michel JJ, Sanghvi K, Rosenbauer J, et al. Type I interferon drives T cell responses to amyloid beta in the central nervous system. Nature Communications. 2026;17:3737. doi:10.1038/s41467-026-72262-6
- Gate D, Saligrama N, Leventhal O, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577:399-404. doi:10.1038/s41586-019-1895-7
- Chen X, Firulyova M, Manis M, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668-677. doi:10.1038/s41586-023-05788-0
- Jorfi M, Park J, Hall CK, et al. Infiltrating CD8+ T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nature Neuroscience. 2023;26:1489-1504. doi:10.1038/s41593-023-01415-3
- Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proceedings of the National Academy of Sciences. 2016;113:E1316-E1325. doi:10.1073/pnas.1525466113
Leave a Reply