
⏱ 9 min read
A 2026 iScience study found that 45 days of clomipramine improved spatial, working, and reference memory in female APP/PS1 Alzheimer’s-model mice, apparently by blocking the E3 ubiquitin ligase Itch rather than by clearing amyloid plaques.1
Research Highlights
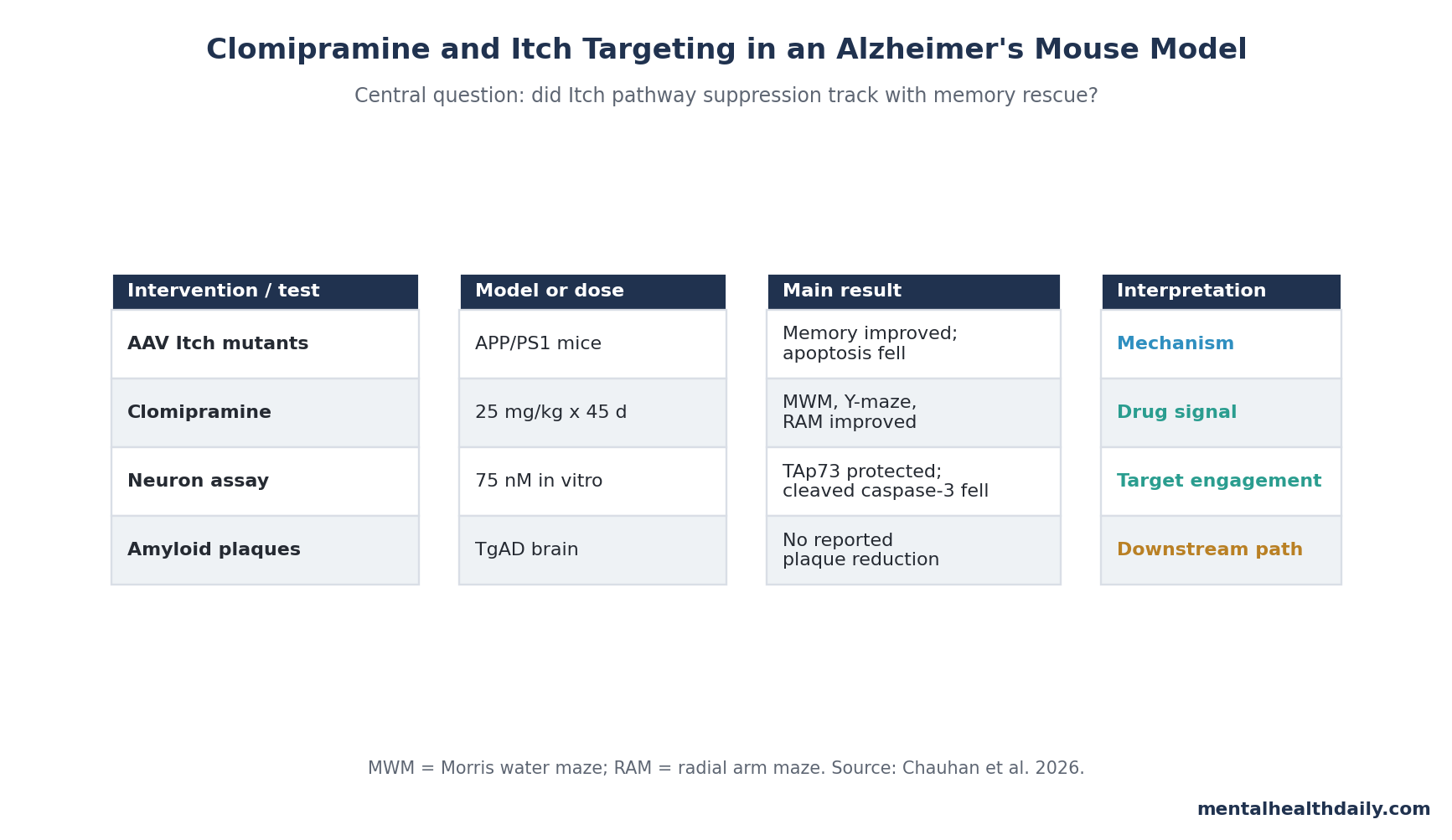
- 45-day clomipramine signal: 6-month female APP/PS1 mice received 25 mg/kg clomipramine every other day for 45 days, then improved across Morris water maze, Y-maze, and 8-arm radial maze testing.1
- Memory rescue was multi-test: final-day water-maze latency improved at p < 0.01, Y-maze alternation improved at p < 0.05 to p < 0.01, and radial-arm-maze errors improved at p < 0.01 to p < 0.001 in groups of 18–19 mice.1
- Itch was the proposed target: clomipramine at 75 nM blocked Aβ42-linked TAp73 ubiquitination in neurons, consistent with direct inhibition of Itch activity rather than a general antidepressant explanation.1
- Apoptosis markers moved with behavior: clomipramine reduced cleaved caspase-3 and PCNA in TgAD cortex, restored TAp73, and lowered TUNEL+/PCNA+ neurons in cortex and hippocampus.1
- Plaques did not explain the effect: amyloid plaque formation was not altered in the treated TgAD mice, so the mouse benefit points downstream of Aβ generation and needs human validation before any clinical leap.1
Clomipramine is a tricyclic antidepressant best known for obsessive-compulsive disorder treatment. In this study, the interesting part was not its serotonin reuptake inhibition; it was clomipramine’s previously reported ability to inhibit Itch, also called AIP4, an E3 ubiquitin ligase that tags proteins for degradation.2
APP/PS1 mice are an Alzheimer’s disease model that overproduces amyloid-β through mutant amyloid precursor protein and presenilin 1 genes. The model is useful for testing amyloid-linked memory biology, but it is still a mouse model: it does not reproduce the full human syndrome, and a memory rescue in these animals is not a dementia treatment result.
Itch Links Amyloid Stress to Neuronal Cell-Cycle Re-Entry
Adult neurons are normally post-mitotic, meaning they do not divide like skin or immune cells. Cell-cycle re-entry describes a damaged state in which neurons begin turning on division-related machinery again; in Alzheimer’s models, that attempted re-entry can lead toward apoptosis, or programmed cell death.3
Pathway chain: Aβ42 stress can hyperactivate the JNK pathway, which then modifies Itch at T222/S232 and K393.4
That aberrant Itch activity promotes degradation of TAp73, a protein involved in neuronal survival, and pushes neurons toward cell-cycle re-entry and apoptosis.
The study tested 2 ways to interrupt that chain:
- Gene therapy approach: adeno-associated virus 9 delivered loss-of-function Itch mutants, T222A/S232A and K393R, into frontal cortex.
- Drug repurposing approach: clomipramine was used as a small-molecule Itch inhibitor after prior screening identified antidepressant drugs as HECT E3 ligase Itch regulators.2
The 2 arms matter because they reduce the chance that the behavioral result is only a generic clomipramine effect. If blocking Itch genetically and pharmacologically both moves memory and apoptosis in the same direction, the mechanism becomes more credible.
Itch Loss-of-Function Mutants Improved Spatial and Working Memory
Researchers injected AAV constructs into the frontal cortex of 6-month WT and APP/PS1 TgAD mice, then waited 45 days before behavioral testing. The frontal-cortex target was mechanistically deliberate: earlier work had found Itch-linked cell-cycle re-entry in cortical neurons, and cortical regions contribute to working-memory and cortex-hippocampus network regulation.1
In the Morris water maze, TgAD mice took longer than WT mice to find the hidden platform. Control AAV did not fix that deficit, and WT Itch did not help. Both loss-of-function Itch mutants significantly reduced escape latency and increased target-quadrant platform crossings in the probe test, with group sizes ranging from 5 to 10 mice.1
Y-maze testing moved in the same direction. TgAD mice showed reduced spontaneous alternation, a working-memory readout. T222A/S232A and K393R Itch mutants significantly increased alternation without changing total arm entries, which argues against a simple locomotor explanation.
Mechanistic readout: cortical sections from TgAD mice had more neurons staining for both TUNEL and PCNA, a combination consistent with apoptosis plus abnormal cell-cycle re-entry. Both Itch mutants reduced that TUNEL+/PCNA+ neuronal population at p < 0.0001 in the histology analysis.1
Clomipramine Reproduced the Itch-Blocking Pattern
Before treating mice, the researchers tested clomipramine in cortical neurons exposed to Aβ42. A 75 nM concentration restored TAp73 and reduced cleaved caspase-3 without the higher-dose toxicity seen at larger concentrations. Immunoprecipitation experiments showed less ubiquitinated TAp73 after clomipramine exposure, fitting the claim that the drug inhibited Itch activity in neurons.1
Mouse dosing then used intraperitoneal clomipramine at 25 mg/kg every other day for 45 days. In the Morris water maze, saline-treated TgAD mice were slower than WT mice, while clomipramine-treated TgAD mice improved until their final-day latency was almost similar to WT mice. Probe testing also improved: treated TgAD mice crossed the target area more often than untreated TgAD mice.
Working and reference memory improved in 2 additional tests. Y-maze spontaneous alternation rose in clomipramine-treated TgAD mice, and radial-arm-maze working-memory and reference-memory errors both decreased. Body weight, swim speed, travel distance, and locomotor measures stayed broadly similar across groups, which weakens a sedation-or-motor-speed explanation for the memory result.1
Amyloid Plaques Stayed Out of the Main Explanation
Plaque-independent mechanism: clomipramine did not alter amyloid plaque formation in TgAD mouse brain. That makes the result different from anti-amyloid antibody logic.
The proposed pathway is downstream of Aβ42 generation: amyloid stress hyperactivates Itch, Itch degrades TAp73, neurons re-enter cell-cycle machinery, apoptosis rises, and cognition worsens.
Why the distinction helps: current Alzheimer’s drug development has leaned heavily on amyloid-targeting strategies, and plaque clearance has not erased the need for safer, complementary mechanisms.5,6
Why it can mislead: plaque-independent mouse rescue can fail in humans for many reasons: dosing, disease stage, blood-brain exposure, anticholinergic burden, and mismatch between APP/PS1 biology and late-life dementia.
Evidence strength: strong for a coherent mouse-and-neuron mechanism, very weak for clinical efficacy. The 2026 study can support “Itch inhibition deserves more Alzheimer’s-model testing.” It cannot support “clomipramine treats Alzheimer’s disease.”
Clomipramine’s Dementia Problem Is Anticholinergic Burden
Repurposing an old psychiatric drug is not automatically easier than inventing a new one. Anticholinergic effects mean a drug blocks acetylcholine signaling, which can worsen confusion, constipation, urinary retention, dry mouth, falls, and cognition in vulnerable older adults. Tricyclic antidepressants, including clomipramine, are usually treated cautiously in geriatric medicine for that reason.
That safety issue does not erase the biology. It changes the development path. The most practical next step may not be giving standard clomipramine to older adults with dementia; it may be testing Itch-selective compounds, lower-exposure regimens, or chemistry that preserves Itch inhibition while reducing anticholinergic liability.
Rossi et al. identified clomipramine and related antidepressant drugs as Itch inhibitors in a high-throughput screen, but a screening hit is not the same as an optimized Alzheimer’s therapy.2 Chauhan et al. strengthened the Itch rationale by showing behavior and apoptosis movement in vivo; drug-development work still has to separate target biology from clomipramine’s broad pharmacology.
Where the Itch Pathway Fits in Alzheimer’s Research
The Itch result sits beside a broader cell-cycle hypothesis of neurodegeneration. Herrup argued that ectopic cell-cycle events are not incidental debris in Alzheimer’s disease; they may be part of the pathologic route into neuronal death.3 Earlier comparative mouse-model work also found that cell-cycle events can mark neurons at risk in Alzheimer’s models.7
Chauhan et al. added target specificity to that framework. Instead of saying cell-cycle re-entry is generally bad, the study names a chain: JNK activation, Itch phosphorylation/autoubiquitination, TAp73 degradation, PCNA elevation, cleaved caspase-3, and TUNEL+/PCNA+ neurons. That chain gives medicinal chemistry a targetable node.
The AAV arm is less immediately practical but mechanistically helpful. Gene delivery of Itch loss-of-function mutants showed that disabling the relevant Itch activation sites improved memory in the same model. The clomipramine arm then showed that a drug-like intervention could push the pathway in a similar direction.
Limits of This Mouse Study
Animal-only design: all behavioral efficacy data came from APP/PS1 mice, not people with mild cognitive impairment or Alzheimer’s dementia. Mouse memory tasks do not translate cleanly into human cognition.
Sex and model specificity: behavioral experiments used female mice because this APP/PS1 model shows a stronger female phenotype. That choice improves signal detection inside the model but leaves male mice, other Alzheimer’s models, and sporadic late-life disease less directly tested.
Brain-region specificity: AAV experiments targeted frontal cortex, while human Alzheimer’s disease involves distributed cortical, hippocampal, vascular, inflammatory, and tau-related pathology. A frontal-cortex rescue in mice is not a full-brain therapy.
Clinical pharmacology: clomipramine has serotonin reuptake, norepinephrine reuptake, anticholinergic, antihistaminic, and other receptor effects. The paper argues for Itch inhibition, but human trials would need dose, exposure, safety, and biomarker evidence that the target is being engaged without unacceptable cognitive side effects.
Questions About Clomipramine and Itch in Alzheimer’s Models
Does this mean clomipramine should be used for Alzheimer’s disease?
No. The data are preclinical. Clomipramine improved memory in APP/PS1 mice and reduced neuronal apoptosis markers, but no randomized human Alzheimer’s trial showed cognitive benefit.
Why is Itch relevant to memory?
Itch is an E3 ubiquitin ligase. In this model, Aβ42 stress appeared to hyperactivate Itch, leading to degradation of TAp73 and abnormal cell-cycle re-entry in neurons. Blocking that chain reduced apoptosis markers and improved mouse memory tasks.
Did clomipramine clear amyloid plaques?
No. The researchers reported no change in amyloid plaque formation. The proposed benefit is downstream of amyloid generation, not plaque clearance.
Why test an antidepressant in an Alzheimer’s mouse model?
Clomipramine had been identified as an Itch inhibitor in prior drug-screening work. The Alzheimer’s experiment tested that target biology, not the idea that treating mood symptoms would reverse dementia.
What would make this clinically convincing?
A convincing human program would need an Itch-engagement biomarker, geriatric safety data, cognitive and functional endpoints, careful anticholinergic monitoring, and comparison against placebo or a better-tolerated Itch-selective compound.
References
- Targeting of Itch by clomipramine or gene therapy improves cognitive defects related to Alzheimer’s disease. Chauhan M et al. iScience. 2026;29:115181. doi:10.1016/j.isci.2026.115181
- High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Rossi M et al. Cell Death & Disease. 2014;5:e1203. doi:10.1038/cddis.2014.113
- The involvement of cell cycle events in the pathogenesis of Alzheimer’s disease. Herrup K. Alzheimer’s Research & Therapy. 2010;2:13. doi:10.1186/alzrt37
- Aberrant activation of neuronal cell cycle caused by dysregulation of ubiquitin ligase Itch results in neurodegeneration. Chauhan M et al. Cell Death & Disease. 2020;11:441. doi:10.1038/s41419-020-2647-1
- Lecanemab in Early Alzheimer’s Disease. van Dyck CH et al. New England Journal of Medicine. 2023;388:9-21. doi:10.1056/NEJMoa2212948
- Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. Sims JR et al. JAMA. 2023;330:512-527. doi:10.1001/jama.2023.13239
- A comparative study of five mouse models of Alzheimer’s disease: cell cycle events reveal new insights into neurons at risk for death. Li L et al. International Journal of Alzheimer’s Disease. 2011;2011:171464. doi:10.4061/2011/171464
Leave a Reply